GOODMAN: Systematic Replication May Make Many Mistakes
Replication seems a sensible way to assess whether a scientific result is right. The intuition is clear: if a result is right, you should get a significant result when repeating the work; if it it’s wrong, the result should be non-significant. I test this intuition across a range of conditions using simulation. For exact replications, the intuition is dead on, but when replicas diverge from the original studies, error rates increase rapidly. Even for the exact case, false negative rates are high for small effects unless the samples are large. These results bode ill for large, systematic replication efforts, which typically prioritize uniformity over fidelity and limit sample sizes to run lots of studies at reasonable cost.
INTRODUCTION
The basic replication rationale goes something like this: (1) many published papers are wrong; (2) this is a serious problem the community must fix; and (3) systematic replication is an effective solution. (In recent months, I’ve seen an uptick in pre-registration as another solution. That’s a topic for another day.) In this post, I focus on the third point and ask: viewed as a statistical test, how well does systematic replication work; how well does it tell the difference between valid and invalid results?
I consider a basic replication scheme in which each original study is repeated once. This is like RPP and EERP, but unlike Many Lab as published which repeated each study 36 times and SSRP which used a two-stage replication strategy. I imagine that the replicators are trying to closely match the original study (direct replication) while doing the replications in a uniform fashion for cost and logistical reasons.
My test for replication success is the same as SSRP (what they call the statistical significance criterion): a replication succeeds if the replica has a significant effect in the same direction as the original.
A replication is exact if the two studies are sampling the same population. This is an obvious replication scenario. You have a study you think may be wrong; to check it out, you repeat the study, taking care to ensure that the replica closely matches the original. Think cold fusion. A replication is near-exact if the populations differ slightly. This is probably what systematic replication achieves, since the need for uniformity reduces precision.
Significance testing of the replica (more precisely, the statistical significance criterion) works as expected for exact replications, but error rates increase rapidly as the populations diverge. This isn’t surprising when you think about it: we’re using the replica to draw inferences about the original study; it stands to reason this will only work if the two studies are very similar.
Under conditions that may be typical in systematic replication projects, the rate of false positive mistakes calculated in this post ranges from 1-71% and false negative mistakes from 0-85%. This enormous range results from the cumulative effect of multiple unknown, hard-to-estimate parameters.
My results suggest that we should adjust our expectations for systematic replication projects. These projects may make a lot of mistakes; we should take their replication failure rates with a grain of salt.
The software supporting this post is open source and freely available in GitHub.
SCENARIO
The software simulates studies across a range of conditions, combines pairs of studies into pairwise replications, calculates which replications pass the test, and finally computes false positive and false negative rates for conditions of interest.
The studies are simple two group comparisons parameterized by sample size and population effect size dpop (dpop ≥ 0). For each study, I generate two groups of n random numbers. One group comes from a standard normal distribution with mean = 0; the other is standard normal with mean = dpop. I then calculate the p-value from a t-test. When I need to be pedantic, I use the term study set for the ensemble of studies for a given combination of n and dpop.
The program varies n from 20 to 500 and dpop from 0 to 1 with 11 discrete values each (a total of 112 = 121 combinations). It simulates 104 studies for each combination yielding about 1.2 million simulated studies. An important limitation is that all population effect sizes are equally likely within the range studied. I don’t consider publication bias which may make smaller effect sizes more likely, or any prior knowledge of expected effect sizes.
To generate pairwise replications, I consider all (ordered) pairs of study sets. For each pair, the software permutes the studies of each set, then combines the studies row-by-row. This multiplies out to 1212 = 14,641 pairs of study sets and almost 150 million simulated replications. The first study of the pair is the original and the second the replica. I consistently use the suffixes 1 and 2 to denote the original and replica respectively.
Four variables parameterize each pairwise replication: n1, n2, d1pop, and d2pop. These are the sample and population effect sizes for the two studies.
After forming the pairwise replications, the program discards replications for which the original study isn’t significant. This reflects the standard practice that non-significant findings aren’t published and thus aren’t candidates for systematic replication.
Next the program determines which replications should pass the replication test and which do pass the test. The ones that should pass are ones where the original study is a true positive, i.e., d1pop ≠ 0. The ones that do pass are ones where the replica has a significant p-value and effect size in the same direction as the original.
A false positive replication is one where the original study is a false positive (d1pop = 0) yet the replication passes the test. A false negative replication is one where the original study is a true positive (d1pop ≠ 0), yet the replication fails the test. The program calculates false positive and false negative rates (abbr. FPR and FNR) relative to the number of replications in which the original study is significant.
My definition of which replications should pass depends only on the original study. A replication in which the original study is a false positive and the replica study a true positive counts as a false positive replication. This makes sense if the overarching goal is to validate the original study. If the goal were to test the result of the original study rather than the study itself, it would make sense to count this case as correct.
To get “mistake rates” I need one more parameter: , the proportion of replications that are true. This is the issue raised in Ioannidis’s famous paper, “Why most published research findings are false” and many other papers and blog posts including one by me. The terminology for “mistake rates” varies by author. I use terminology adapted from Jager and Leek. The replication-wise false positive rate (RWFPR) is the fraction of positive results that are false positives; the replication-wise false negative rate (RWFNR) is the fraction of negative results that are false negatives.
RESULTS
Exact replications
A replication is exact if the two studies are sampling the same population; this means d1pop = d2pop.
Figure 1 shows FPR for n1 = 20 and n2 varying from 50 to 500. The x-axis shows all four parameters using d1, d2 as shorthand for d1pop, d2pop. d1pop = d2pop = 0 throughout because this is the only way to get false positives with exact replications. Figure 2 shows FNR for the same values of n1 and n2 but with d1pop = d2pop ranging from 0.1 to 1.
I mark the conventionally accepted thresholds for false positive and negative error rates (0.05 and 0.2, resp.) as known landmarks to help interpret the results. I do not claim these are the right thresholds for replications.
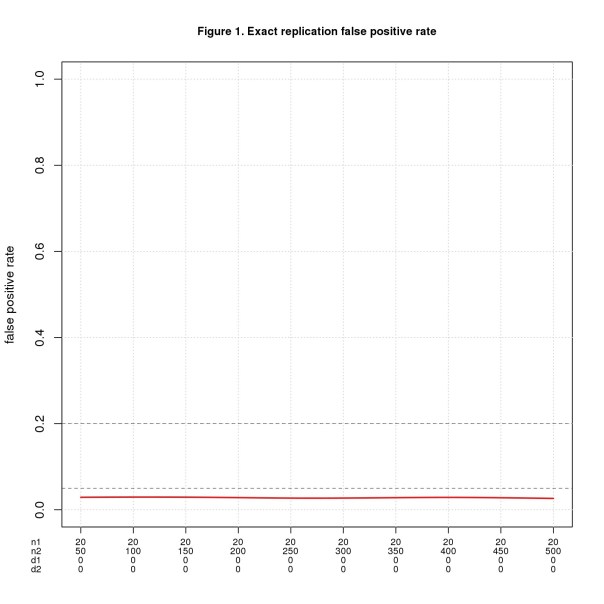

For this ideal case, replication works exactly as intuition predicts. FPR is the significance level divided by 2 (the factor of 2 because the effect sizes must have the same direction). Theory tell us that FNR = 1 – power and though not obvious from the graph, the simulated data agrees well.
As one would expect, if the population effect size is small, n2 must be large to reliably yield a positive result. For d = 0.2, n2 must be almost 400 in theory and 442 in the simulation to achieve FNR = 0.2; to hit FNR = 0.05, n2 must be more than 650 (in theory). These seem like big numbers for a systematic replication project that needs to run many studies.
Near exact replications
A replication is near-exact if the populations differ slightly, which means d1pop and d2pop differ by a small amount, near; technically, abs(d1pop – d2pop) ≤ near.
I don’t know what value of near is reasonable for a systematic replication project. I imagine it varies by research area depending on the technical difficulty of the experiments and the variability of the phenomena. The range 0.1-0.3 feels reasonable. I extend the range by 0.1 on each end just to be safe.
Figure 3 uses the same values of n1, n2, and d1pop as Figure 1, namely n1 = 20, n2 varies from 50 to 500, and d1pop = 0. Figure 4 uses the same values of n1 and n2 as Figure 2 but fixes d1pop = 0.5, a medium effect size. In both figures, d2pop ranges from d1pop – near to d1pop + near with values less than 0 or greater than 1 discarded. I restrict values to the interval [0,1] because that’s the range of d in the simulation.
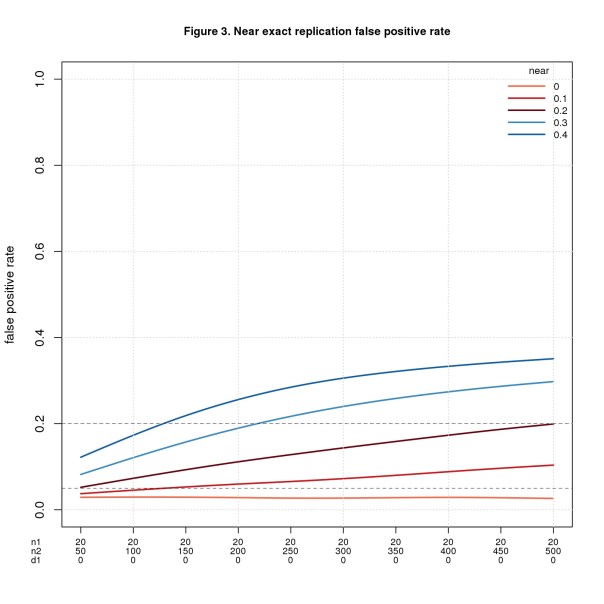

FPR is fine when n2 is small, esp. when near is also small, but gets worse as n2 (and near) increase. It may seem odd that the error rate increases as the sample size increases. What’s going on is a consequence of power. More power is usually good, but in this setting every positive is a false positive, so more power is bad. This odd result is a consequence of how I define correctness. When the original study is a false positive (d1pop = 0) and the replica a true positive (d2pop ≠ 0), I consider the replication to be a false positive. This makes sense if we’re trying to validate the original study. If instead we’re testing the result of the original study, it would make sense to count this case as correct.
FNR behaves in the opposite direction: bad when n2 is small and better as n2 increases.
To show the tradeoff between FPR and FNR, Figure 5 plots both error rates for near = 0.1 and near = 0.3.

For near =0.1, n2 = 150 is a sweet spot with both error rates about 0.05. For near = 0.3, the crossover point is n2 = 137 with error rates of about 0.15.
FNR also depends on d1pop for “true” cases, i.e., when the original study is a true positive, getting worse when d1pop is smaller and better when d1pop is bigger. The table below shows the error rates for a few values of n2, near, and d1pop. Note that FPR only depends on n2 and near, while FNR depends on all three parameters. The FNR columns are for different values of d1pop in true cases.

FNR is great for d1pop = 0.8, mostly fine for d1pop = 0.5, and bad for d1pop = 0.2. Pushing up n2 helps but even when n2 = 450, FNR is probably unacceptable for d1pop = 0.2. Increasing n2 worsens FPR. It seems the crossover point above, n2 = 137, is about right. Rounding up to 150 seems a reasonable rule-of-thumb.
Replication-wise error rates
The error rates reported so far depend on whether the original study is a false or true positive: FPR assumes the original study is a false positive, FNR assumes it’s a true positive. The next step is to convert these into replication-wise error rates: RWFPR and RWFNR. To do so, we need one more parameter: prop.true, the proportion of replications that are true.
Of course, we don’t know the value of prop.true; arguably it’s the most important parameter that systematic replication is trying to estimate. Like near , it probably varies by research field and may also depend on the quality of the investigator. Some authors assume prop.true = 0.5, but I see little evidence to support any particular value. It’s easy enough to run a range of values and see how prop.true affects the error rates.
The table below shows the results for near = 0.1, 0.3 as above, and prop.true ranging from 0.1 to 0.9. The RWFPR and RWFNR columns are for different values of d1pop in “true” cases, i.e., when the original study is a true positive.
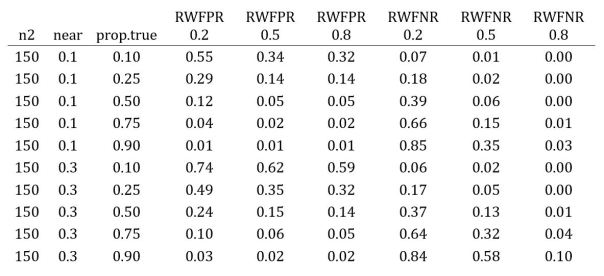
Check out the top and bottom rows. The top row depicts a scenario where most replications are false (prop.true = 0.1) and the replicas closely match the original studies (near = 0.1); for this case, most positives are mistakes and most negatives are accurate. The bottom row is a case where most replications are true (prop.true = 0.9) and the replicas diverge from the originals (near = 0.3); here most positives are correct and, unless d1pop is large, most negatives are mistakes.
Which scenario is realistic? There are plenty of opinions but scant evidence. Your guess is as good as mine.
DISCUSSION
Systematic replication is a poor statistical test when used to validate published studies. Replication works well when care is taken to ensure the replica closely matches the original study. This is the norm in focused, one-off replication studies aiming to confirm or refute a single finding. It seems unrealistic in systematic replication projects, which typically prioritize uniformity over fidelity to run lots of studies at reasonable cost. If the studies differ, as they almost certainly must in systematic projects, mistake rates grow and may be unacceptably high under many conditions.
My conclusions depend on the definition of replication correctness, i.e., which replications should pass. The definition I use in this post depends only on the original study: a replication should pass if the original study is a true positive; the replica study is just a proxy for the original one. This makes sense if the goal is to validate the original study. If the goal were to test the result of the original study rather than the study itself, it would make sense to let true positive replicas count as true positive replications. That would greatly reduce the false positive rates I report.
My conclusions also depend on details of the simulation. An important caveat is that population effect sizes are uniformly distributed across the range studied. I don’t consider publication bias which may make smaller effect sizes more likely, or any prior knowledge of expected effect sizes. Also, in the near exact case, I assume that replica effect sizes can be smaller or larger than the original effect sizes; many investigators believe that replica effect sizes are usually smaller.
My results suggest that systematic replication is unsuitable for validating existing studies. An alternative is to switch gears and focus on generalizability. This would change the mindset of replication researchers more than the actual work. Instead of trying to refute a study, you would assume the study is correct within the limited setting of the original investigation and try to extend it to other settings. The scientific challenge would become defining good “other settings” – presumably there are many sensible choices — and selecting studies that are a good fit for each. This seems a worthy problem in its own right that would move the field forward no matter how many original studies successfully generalize.
I’ve seen plenty of bad science up close and personal, but in my experience statistics isn’t the main culprit. The big problem I see is faulty research methods. Every scientific field has accepted standard research methods. If the methods are bad, even “good” results are likely to be wrong; the results may be highly replicable but wrong nonetheless.
The quest to root out bad science is noble but ultimately futile. “Quixotic” comes to mind. Powerful economic forces shape the size and make-up of research areas. Inevitably some scientists are better researchers than others. But “Publish or Perish” demands that all scientists publish research papers. Those who can, publish good science; those who can’t, do the best they can.
We will do more good by helping good scientists do good science than by trying to slow down the bad ones. The truly noble quest is to develop tools and techniques that make good scientists more productive. That’s the best way to get more good science into the literature.
Nat Goodman is a retired computer scientist living in Seattle Washington. His working years were split between mainstream CS and bioinformatics and orthogonally between academia and industry. As a retiree, he’s working on whatever interests him, stopping from time-to-time to write papers and posts on aspects that might interest others. He can be contacted at natg@shore.net.

Surely we cannot expect results to replicate while we continue to use p = 0.05 to indicate “statistical significance”.
It still seems to be very common to confuse the type 1 error rate with the false positive risk. But if you observe a p value close to 0.05 and declare that you’ve made a discovery, there is a 20 -30% risk (depending on how exactly you calculate it) that you are wrong -it is a false positive. And that is the result for a plausible hypothesis (prior = 0.05). For implausible hypotheses, the risk of false positives would be still higher. More details at https://arxiv.org/abs/1802.04888
LikeLike
Thanks, David, for the interesting comment! I hope it’s clear in this post that I distinguished the type 1 error rate (“false positive rate” in the post) from “mistake rate”: what you call “false positive risk” and I call “replication-wise false positive rate” in the post.
I think the terminology and acronyms contribute to the confusion. Upon reflection, perhaps it would be more clear if we reverted to the old terminology of “positive and negative predictive value”. The acronyms (PPV, NPV) are distinctive and well-known, and the concepts perhaps closer to what really matter to scientists, namely given a positive (resp. negative) finding, what’s the probability that the finding is a true positive (resp. negative) result.
Do you think this would help?
LikeLike